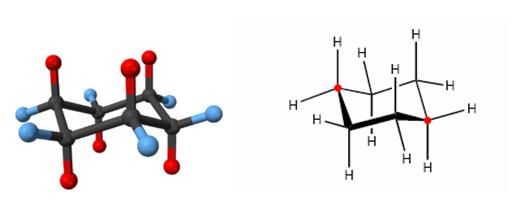
- Cicloalcanos superiores y alcanos policíclicos:
El ciclohexano es especial porque la forma de su estructura silla es adecuada a la estructura tetraédrica del carbono y carece de tensión angular y por eclipsamiento H/H. Los cicloalcanos superiores carecen de esa simetría y contienen algo de tensión, hasta el ciclotetradecano que puede adoptar la conformación zig-zag típica de los alcanos lineales.
Ciclotetradecano.
Cicloalcanos policíclicos
Cicloalcanos fusionados
 |
| La decalina es en realidad como dos ciclohexanos sustituídos en 1,2. Por tanto tiene dos isómeros: |

Tiene dos conformaciones degeneradas en equilibrio |

Sólo presenta una conformación. La otra tendría los dos metilenos en axial, lo que crearía una tensión enorme al estar esos dos metilenos incluídos en otro anillo. |
Cicloalcanos puenteados

El norbornano puede considerarse como dos ciclopentanos que tienen tres carbonos comunes o como un ciclohexano que tiene un puente metileno en posiciones 1,4. La molécula es muy rígida, no puede adoptar una conformación silla y tiene tensión de anillo y de eclipsamiento. |
-el núcleo de los esteroides:
Los esteroides son derivados del núcleo del ciclopentanoperhidrofenantreno o esterano que se compone de carbono e hidrógeno formando cuatro anillos fusionados, tres hexagonales y uno pentagonal; posee 17 átomos de carbono. En los esteroides esta estructura básica se modifica por adición de diversos grupos funcionales, como carbonilos e hidroxilos (hidrófilos) o cadenas hidrocarbonadas (hidrófobas).
- Reacciones de los alcanos:
Las reacciones más importantes de los alcanos son la pirólisis,la combustión y la halogenación.

-Oxidación y combustión:
proceso de oxidación rápida o quema de una sustancia con evolución simultánea de calor y, por lo general, luz. En el caso de combustibles comunes, el proceso es una de combinación química con el oxígeno atmosférico para producir productos principales como el dióxido de carbóno, el monóxido de carbóno, y el agua, juntos con productos como el dióxido de azufre que puede ser generado por los componentes menores del combustible. El término de combustión, sin embargo, también abraza la oxidación en el amplio sentido químico, y el agente que se oxida puede ser el ácido nítrico, ciertos percloratos, o hasta el cloro o el flúor.

- Petróleo y craqueo de hidrocarburos: Es un líquido oleoso bituminoso de origen natural compuesto por diferentes sustancias orgánicas. Se encuentra en grandes cantidades bajo la superficie terrestre y se emplea como combustible y materia prima para la industria química. El petróleo y sus derivados se emplean para fabricar medicinas, fertilizantes, productos alimenticios, objetos de plástico, materiales de construcción, pinturas o textiles y para generar electricidad. El proceso de craqueo térmico, o pirólisis a presión, se desarrolló en un esfuerzo para aumentar el rendimiento de la destilación. En este proceso, las partes más pesadas del crudo se calientan a altas temperaturas bajo presión. Esto divide (craquea) las moléculas grandes de hidrocarburos en moléculas más pequeñas, lo que aumenta la cantidad de gasolina —compuesta por este tipo de moléculas— producida a partir de un barril de crudo. No obstante, la eficiencia del proceso era limitada, porque debido a las elevadas temperaturas y presiones se depositaba una gran cantidad de coque (combustible sólido y poroso) en los reactores. Esto, a su vez, exigía emplear temperaturas y presiones aún más altas para craquear el crudo. Más tarde se inventó un proceso de coquefacción en el que se recirculaban los fluidos; el proceso funcionaba durante un tiempo mucho mayor con una acumulación de coque bastante menor. Muchos refinadores adoptaron este proceso de pirólisis a presión.

Halogenación de alcanos: mecanismo:
La halogenación es el proceso químico mediante el cual se adicionan uno o varios átomos de elementos del grupo de los halógenos (grupo 17 de la tabla periódica) a una molécula orgánica. Una de las halogenaciones más simples es la halogenación de alcanos. En estas reacciones los átomos de hidrógeno de los alcanos resultan sustituidos total o parcialmente por átomos del grupo de los halógenos. La reacción que tiene lugar es la siguiente:

Son posibles una gran variedad de productos químicos. La composición de la mezcla de productos vendrá dada por la concentración de los reactivos y otras condiciones del medio de reacción, por ejemplo, la temperatura. En una halogenación se incorpora un átomo de halógeno a una molécula. Existen descripciones más concretas que especifican el tipo de halógeno: fluoración, cloración, bromación y yodación. Existen varios tipos principales de halogenación, incluyendo:

- Fuerza de los enlaces de los alcanos:
Las aplicaciones de los alcanos pueden ser determinadas bastante bien de acuerdo al número de átomos de carbono. Los cuatro primeros alcanos son usados principalmente para propósitos de calefacción y cocina, y en algunos países para generación de electricidad. El metano y el
etano
son los principales componentes del gas natural; pueden ser almacenados como gases bajo presión. Sin embargo, es más fácil transportarlos como líquidos: esto requiere tanto la compresión como el enfriamiento del gas.
El propano y el butano pueden ser líquidos a presiones moderadamente bajas y son conocidos como gases licuados del petróleo (GLP). Por ejemplo, el propano se usa en el quemador de gas propano, el butano en los encendedores descartables de cigarrillos. Estos dos alcanos son usados también como propelentes en
pulverizadores
.

- Estructura y estabilidad de los radicales alquilo:
Por definición, la energía de disociación de enlace es la cantidad de energía requerida para convertir un mol de alcano en radicales y átomos de hidrógeno. Podemos apreciar que la energía requerida para la formación de los distintos tipos de radicales decrece en el orden: CH3. > 1º > 2º > 3º.
Si se necesita menos energía para formar un radical que otro, esto sólo puede significar que, en relación con el alcano que lo origina, una radical contiene menos energía que el otro, es decir, es más estable.
No intentamos comparar el contenido energético absoluto de los radicales metilo o etilo, por ejemplo; simplemente observamos que la diferencia en energía entre el metano y los radicales metilo e mayor que la diferencia entre el etano y los radicales etilo. Cuando comparamos estabilidades relativas de radicales libre, debe entenderse que nuestro patrón para cada radical es el alcano del cual se deriva. Como veremos, es precisamente ésta la clase de estabilidad que nos interesa.
Entonces, en relación con el alcano del cual se forma, el orden de estabilidad de los radicales libres es:
Estabilidad de radicales:
3º > 2º > 1º > CH3.
- Aspectos sintéticos de las reacciones radicalarias:
Los intermedios de reacción son especies de tiempo de vida media corto y no están presentes nunca en altas concentraciones debido a que reaccionan tan rápidamente como se forman. Las especies de carbono trivalente (tres enlaces) se clasifican de acuerdo con su carga, que depende del número de electrones no compartidos. Los carbocationes, o iones carbonio tienen todos los electrones externos compartidos y están cargados positivamente.
Los radicales, también llamados radicales libres, tienen un electrón no compartido y son neutros.
Los carbaniones tienen un numero par de electrones no compartidos y tienen carga negativa. Estructura y estabilidad de los carbocationes.
Un carbocatión tiene hibridación sp2 con una estructura plana y ángulos de enlace de 120°.

4.Estereoisomeria
-Moléculas quirales:
es la propiedad que tienen ciertas moléculas de poder existir bajo dos formas que son imágenes especulares la una de la otra, es decir, una es la imagen reflejada en un espejo de la otra. Muchos objetos creados por el ser humano tienen esa misma propiedad, como los guantes, las escaleras de caracol o las sillas con pala para escribir.
Las moléculas quirales se diferencian de las aquirales en que tienen actividad óptica, desvían el plano en el que vibra la luz polarizada. Una de las formas lo desvía a la derecha y la otra a la izquierda. Por lo demás, ambas formas corresponden al mismo compuesto y tienen idénticas propiedades.
El interés de la Ciencia por estas moléculas sería prácticamente anecdótico de no ser por el hecho de que el grupo de compuestos más amplio y característico de los que forma la materia viva, responsable de la gran mayoría de los fenómenos vitales, las proteínas, está compuesto por moléculas quirales, los aminoácidos. Además, de las dos posibles formas que pueden existir de cada aminoácido, en las proteínas sólo aparece una y siempre la misma sea cual sea la proteína y en cualquier ser vivo.
Las consecuencias de este hecho, cuya explicación constituye uno de los grandes enigmas actuales de la Ciencia, se vuelven imprevisibles cuando un ser vivo debe enfrentarse a una molécula que tiene la forma opuesta a la que existe en la Naturaleza, lo cual ocurre siempre que el compuesto se produce de manera artificial, se forman a partes iguales las dos versiones moleculares siendo extremadamente difícil separar unas de otras. El método que usó el primer científico que hizo esta separación es buena prueba de ello.
- Propiedades físicas de los enantiómeros:
En química se dice que dos estereoisómeros son enantiómeros si la imagen especular de uno no puede ser superpuesta con la del otro. Dicho de otra forma: un enantiómero es una imagen especular no superponible de sí mismo. Tienen las mismas propiedades físicas y químicas, excepto por la interacción con el plano de la luz polarizada o con otras moléculas quirales. Son moléculas quirales. La mezcla en cantidades equimolares de cada enantiómero en una solución se denomina
mezcla racémica
y es ópticamente inactiva.

-Actividad óptica:
La rotación óptica o actividad óptica es la rotación de la polarización lineal de la luz cuando viaja a través de ciertos materiales. Suele ser un fenómeno que ocurre en soluciones que presentan moléculas quirales tales como la sacarosa (azúcar), sólidos con planos cristalinos rotados, tales como el cuarzo, y la polarización circular de gases atómicos o moleculares. Se emplea en la industria de elaboración de azúcar para medir en los siropes la concentración de azúcares, en óptica para manipular la polarización, en química para caracterizar sustancias en solución acuosa, y en medicina está siendo evaluado en la actualidad como un método de determinación de la concentración de azúcar en sangre en casos de personas que sufren la
diabetes
.

- Racematos:
Esquemáticamente, el proceso es el siguiente:
- Centros estereogénicos:
Un centro estereogénico es un átomo unido a varios otros átomos que tienen la propiedad de que si se intercambian dos de las uniones da lugar a diferentes estereoisómeros ( no necesariamente enantiómeros).

-Nomenclatura de los enantiómeros: La regla R-S:
Para dar notación R/S a un centro quiral es necesario asignar prioridades a los sustituyentes mediante las siguientes reglas:
Las prioridades de los átomos unidos al quiral se dan por números atómicos. En el caso de isótopos, tiene prioridad el de mayor masa atómica.
Cuando dos o más sustituyentes unidos al centro quiral tengan la misma prioridad, se continua comparando las cadenas átomo a átomo hasta encontrar un punto de diferencia.
Los enlaces dobles y triples se desdoblan considerándolos como si fueran enlaces sencillos.
Para asignar notación R/S seguimos el orden de prioridades a, b, c de los sustituyentes. Si esta sucesión se realiza en el sentido de las agujas del reloj se dice que el centro es R (rectus, latín derecha). Si se sigue el sentido contrario a las agujas al recorrer las prioridades a, b, c se dice que es S (sinester, latín izquierda). Esta regla sólo es válida cuando el grupo d está hacia el fondo del plano (enlace a trazos), si d sale hacia nosotros (cuña) la notación es la contraria (R giro a la izquierda, S giro a la derecha).

- Convenio E-Z para isómeros cis-trans:
La nomenclatura cis-trans para los isómeros geométricos a veces falla, ya que da un nombre ambiguo; por ejemplo, los isómeros del 1-bromo-1-cloropropeno no son claramente cis o trans, ya que no es obvio a qué sustituyentes se refieren como cis o trans. El sistema E-Z de nomenclatura para los isómeros sigue el convenio de Cahn-Ingold-Prelog para los átomos de carbono asimétricos y asigna una única configuración E o Z a cualquier doble enlace que pueda presentar isomería geométrica. Como en el caso de cis y trans, si los grupos más importantes de cada carbono están en el mismo lado del enlace doble, el alqueno tendría una geometría Z. Si están en lados opuestos al enlace doble, la geometría es E.
-Proyecciones de Fischer:
Las proyecciones de Fischer son utilizadas en
química orgánica
para representar en dos dimensiones la disposición espacial de moléculas en las que uno o más átomos de carbono están unidos a 4 sustituyentes diferentes.
Estos átomos pueden existir en dos configuraciones espacialmente diferentes, que son imágenes especulares (simétricas respecto a un plano) entre sí, como lo son la mano derecha de la izquierda, y al igual que éstas no son superponibles en el espacio. Estos átomos constituyen centros quirales o de isomería espacial. Cada centro quiral da lugar a dos moléculas isómeras especulares o
enantiómeros
. Una molécula con 2 centros quirales puede tener 4 estereoisómeros (22 esteroisómeros, 2 parejas de enantiómeros); una con 3 centros quirales puede tener 8 estereoisómeros (23 esteroisómeros, 4 parejas de enantiómeros); y así sucesivamente.

- Compuestos que contienen más de un estereocentro:
-Diastereómeros:
son una clase de estereoisómeros tales, que no son superponibles pero tampoco son imagen especular uno del otro, es decir, no son
enantiómeros
.

- Compuestos meso:
Se denomina compuesto meso a aquel que, conteniendo átomos de carbono asimétricos, es aquiral (existe un plano de simetría). Por lo tanto, su imagen especular es en realidad el mismo compuesto.
Este tipo de compuestos carece de actividad optica a pesar de contener en su estructura centros
estereogénicos.
Los compuestos meso contienen un plano de simetría que divide la molécula en dos, de tal forma que una mitad es la imagen especular de la otra . Las estructuras meso se basan en el número de centros estereogénicos representados por la formula 2^n donde n es el número de centros y dicha fórmula lo va a definir la cantidad de estereoisómeros que tenemos en la molécula.Por ejemplo, uno de los isómeros del ácido tartárico representado abajo es un compuesto meso. Existe un plano especular interno, que biseca la molécula: al rotar la molécula 180° sobre el plano perpendicular a la pantalla, se obtiene la misma estereoquímica. (ver
Proyección de Fischer)
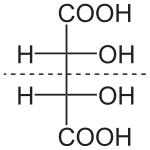
- Estereoisomería en compuestos cíclicos:
La geometria y la naturaleza de la doble ligadura nos da lugar a la presencia de estereoisómeros en los alquenos, esto no se presenta en el caso de los alcanos donde la ligadura sencilla entre C-C permite la rotación libre, o en el caso de los alquinos donde la triple ligadura C-C es lineal y no da lugar a isómeros
Si recordamos la geometría de la doble ligadura, esta es planar y tiene un ángulo de 120 grados entre los sustituyentes. La presencia de la doble ligadura formada por los orbitales p no hibridizados hace que la rotación de esta ligadura tenga una barrera energética muy alta, evitando la libre rotación a condiciones de temperatura ambiente.
Esta estrucutra rígida planar la podemos ver en el caso del etileno
, CH2=CH2, se puede observar que todos los átomos están sobre el mismo plano.
Vamos a definir a los estereoisómeros como compuestos que:
a) Tienen la misma fórmula molecular, o sea cuentan con el mismo número y tipo de átomos.
b) La conectividad entre los átomos es la misma, solo difieren en la orientación espacial de los átomos.
Cuando vimos los compuestos cíclicos se mencionó la presencia de isómeros cis/tran, dependiendo si los sustituyentes estaban del mismo lado del anillo o en lados opuestos.
El estereoisomerismo en el caso de las dobles ligaduras se observa cuando esta se encuentra disustituida en los carbonos no terminales, o hay una atri o tetra sustitución.

Reacciones químicas y estereoisomería:
síntesis estereoselectivas:
Síntesis estereoselectiva de 2’-desoxi-, aciclo-, e isonucleósidos se han desarrollado nuevos métodos para la obtención de análogos de nucleósidos. Más concretamente el trabajo se ha dividido en tres apartados que se desarrollarán a continuación: En primer lugar se ha llevado a cabo un estudio exhaustivo de un nuevo método de síntesis estereoselectiva de 2’-desoxinucleósidos a partir de 1-tio y 1-selenoglicósidos basado en la activación del hidroxilo 2-OH en condiciones de Mitsunobu y que transcurre a través de una transposición 1,2 del sustituyente anomérico seguido de glicosilación. De este estudio se extraen las siguientes conclusiones concretas:
-Cuando se partió de la tetraacetilribosa, la reacción condujo mayoritariamente a los 2-sulfenil y 2-selenenil--D-arabino nucleósidos, obteniéndose los mejores resultados a partir del terc-butil 1-tio-ribo-glicósido con N3-Bz-T (65%, =10:1).
-Cuando se partió del 2,3,5-tri-O-benzil-D-arabino-furanósido de metilo, la reacción condujo a los 2-sulfenil y 2-selenenil--D-ribo nucleósidos, obteniéndose excelentes estereoselectividades con el terc-butil 1-tio-arabino-glicósido (=1:12 con Cl6-purina y=0:1 con N3-Bz-T).
La reacción es altamente estereoselectiva, pero no es estereoespecífica, lo que parece indicar que la reacción no transcurre a través de un intermedio episulfonio o selenonio. La obtención exclusiva del anómero en el caso del terc-butil 1-tio-arabino-glicósido con N3-Bz-T puede ser debida al fuerte impedimento estérico ejercido por el grupo terc-butilo.

– Resolución de una mezcla racémica:
consiste en la separación de los enantioméros mediante la conversión de la mezcla racémica en una mezcla dediastereoisómeros. Para ello, la mezcla de enantiómeros se hace reaccionar con el compuesto quiral que recibe el nombre de agente de resolución; con la cual se transforman los enantiómeros en diasteroisómeros, los cuales se pueden separar mediante destilación, cristalización o
cromatografía. Una vez separados, se procede a la eliminación del agente de resolución para obtener cada uno de los enantiómeros puros.

5. Alqueno y Alquinos
-Clasificación:
Los alquenos son hidrocarburos insaturados que contienen uno o más dobles enlaces C = C. El nombre de los hidrocarburos insaturados se debe a que los carbonos que sostienen el doble enlace todavía pueden enlazarse a hidrógenos.
Clasificación de los alquenos
Alquenos simples: presentan un solo doble enlace
C = C.
Alcadienos :
Vecinos o acumulados: presentan dos dobles enlaces sostenidos del mismo carbono,
C H 2 = C = CH CH 3 .
Extremos: presentan dos dobles enlaces, uno en cada extremo de la molécula,
CH 2 = CH CH 2 CH CH 2 .
Conjugados : presentan dos dobles enlaces, alternados;
CH 2 CH CH CH CH 3 .
Alcapolienos: presentan más de dos dobles enlaces: CH 2 CC CH CH 3
Los alquinos son hidrocarburos alifáticos con al menos un triple enlace entre dos átomos de carbono. Se trata de compuestos metaestables debido a la alta energía del triple
enlace carbono-carbono. Su fórmula general es CnH2n-2v
- Nomenclatura:
Nomenclatura de los alquenos:
- Localizar la cadena continua de carbonos más larga y que contenga la mayor cantidad posible de dobles enlaces. Nombrar esta cadena con las mismas raíces utilizadas para los alcanos; pero dándoles la terminación característica de los alquenos ( eno, dieno, trieno ), según sea el número de dobles enlaces
presentes en la molécula , e indicando la posición de cada uno de ellos.
-
- Numerar los carbonos de la cadena anterior, de tal manera que los dobles enlaces queden en la menor posición posible. Si éstos equidistan de ambos extremos, numerar la cadena de tal manera que las ramificaciones queden en la menor posición posible.
- Nombrar cada una de las ramificaciones de igual manera que en los alcanos.
- Terminar el nombre del alqueno escribiendo el nombre de la cadena principal o base de la molécula.
- Cuando una ramificación presente dobles enlaces, se nombrará utilizando las mismas raíces que para los grupos alquil; pero con la teminación característica de los grupos alqueniles (enil, dienil, trienil) según sea el número de dobles enlaces presentes en la ramificación, e indicando cada una de sus posiciones.
- NOTA: Cuando se presenta el doble enlace es común encontrar isómeros cis-trans. Estos se nombrarán anteponiendo el prefijo cis o trans a todo el nombre del alqueno, según sea el caso.
Para dar nombre a los hidrocarburos del tipo alquino se siguen ciertas reglas similares a las de los
alquenos.
- Se toma como cadena principal la cadena continua más larga que contenga el o los triples enlaces.
- La cadena se numera de forma que los átomos del carbono del triple enlace tengan los números más bajos posibles.
-
- Dicha cadena principal se nombra con la terminación -ino, especificando el número de átomos de carbono de dicha cadena con un prefijo (et- dos, prop- tres, but- cuatro; pent-; hex-; etc). Ej.: propino, CH3-C
 CH.
CH.
- En caso necesario, la posición del triple enlace se indica mediante el menor número que le corresponde a uno de los átomos de carbono del enlace triple. Dicho número se sitúa antes de la terminación -ino. Ej.: CH3-CH2-CH2-CH2-CC-CH3, hept-2-ino.
- Si hay varios triples enlaces, se indica con los prefijos di, tri, tetra... Ej.: octa-1,3,5,7-tetraino, HCC-CC-CC-CCH.
- Si existen dobles y triples enlaces, se da el número más bajo al doble enlace. Ej.: pent-2-en-4-ino, CH3-CH=CH-CCH
- Los sustituyentes tales como átomos de halógeno o grupos
- alquilo se indican mediante su nombre y un número, de la misma forma que para el caso de los alcanos. Ej.: 3-cloropropino, HCC-CH2Cl; 2,5-dimetilhex-3-ino, CH3-C(CH3)-CC-C(CH3)-CH3.

- Modelo orbital de un doble y un triple enlace: enlace p.
Los átomos de carbono también pueden formar entre sí enlaces dobles y triples, denominados insaturaciones. En los enlaces dobles, la hibridación ocurre entre el orbital 2s y dos orbitales 2p, y queda un orbital p sin hibridar. A esta nueva estructura se la representa como:
- 1s² (2sp²)¹ (2sp²)¹ (2sp²)¹ 2pz¹
Al formarse el enlace doble entre dos átomos, cada uno orienta sus tres orbitales híbridos con un ángulo de 120°, como si los dirigieran hacia los vértices de un triángulo equilátero. El orbital no hibridado queda perpendicular al plano de los 3 orbitales sp².
A este doble enlace se lo denomina π (pi), y la separación entre los carbonos se acorta. Este enlace es más débil que el enlace σ (sigma) y, por tanto, más reactivo.
Este tipo de enlace da lugar a la serie de los
alquenos.
El segundo tipo de insaturación es el enlace triple: el carbono hibrida su orbital 2s con un orbital 2p. Los dos orbitales p restantes no se hibridan, y su configuración queda:
- 1s² (2sp)¹ (2sp)¹ 2py¹ 2pz¹
Al formarse el enlace entre dos carbonos, cada uno traslada uno de sus 2 orbitales sp para formar un enlace sigma entre ellos; los dos orbitales p sin hibridar de cada átomo se traslapan formando los dos enlaces (π) restantes de la triple ligadura, y al final el último orbital sp queda con su electrón disponible para formar otro enlace.
A los dos últimos enlaces que formaron la triple ligadura también se les denomina enlaces pi(π), y todo este conjunto queda con ángulos de 180° entre el triple enlace y el orbital sp de cada átomo de carbono, es decir, adquiere una estructura lineal.
La distancia entre estos átomos se acorta más, por lo que es incluso más reactivo que el doble enlace
El segundo tipo de insaturación es el enlace triple: el carbono hibrida su orbital 2s con un orbital 2p. Los dos orbitales p restantes no se hibridan, y su configuración queda:
- 1s² (2sp)¹ (2sp)¹ 2py¹ 2pz¹
Al formarse el enlace entre dos carbonos, cada uno traslada uno de sus 2 orbitales sp para formar un enlace sigma entre ellos; los dos orbitales p sin hibridar de cada átomo se traslapan formando los dos enlaces (π) restantes de la triple ligadura, y al final el último orbital sp queda con su electrón disponible para formar otro enlace.
A los dos últimos enlaces que formaron la triple ligadura también se les denomina enlaces pi(π), y todo este conjunto queda con ángulos de 180° entre el triple enlace y el orbital sp de cada átomo de carbono, es decir, adquiere una estructura lineal.
La distancia entre estos átomos se acorta más, por lo que es incluso más reactivo que el doble enlace

- Propiedades físicas de alquenos:
La presencia del doble enlace modifica ligeramente las propiedades físicas de los alquenos frente a los
alcanos. De ellas, la temperatura de ebullición es la que menos se modifica. La presencia del doble enlace se nota más en aspectos como la polaridad y la acidez.
Polaridad
Dependiendo de la estructura, puede aparecer un momento dipolar débil. El enlace alquilo-alquenilo está polarizado en la dirección del átomo con orbital sp2, ya que la componente s de un orbital sp2 es mayor que en un sp3 (esto podría interpretarse como la proporción de s a p en la molécula, siendo 1:2 en sp2 y 1:3 en sp3, aunque dicha idea es simplemente intuitiva). Esto es debido a que los electrones situados en orbitales híbridos con mayor componente s están más ligados al núcleo que los p, por tanto el orbital sp2 es ligeramente atrayente de electrones y aparece una polarización neta hacia él. Una vez que tenemos polaridad en el enlace neta, la geometría de la molécula debe permitir que aparezca un momento dipolar neto en la molécula, como se aprecia en la figura
inferior. 
'La primera molécula' es
cis y tenemos un momento dipolar neto, pero la segunda trans, pese a tener dos enlaces ligeramente polarizados el momento dipolar neto es nulo al anularse ambos momentos dipolares.
Acidez
El carbono alquenílico tiene mayor acidez frente a los alcanos, debido también a la polaridad del enlace. Así, el etano (alcano) tiene un pKa de 50 (ó un Ka de 10-50) frente al pKa = 44 del eteno. Este hecho se explica fácilmente considerando que, al desprenderse un electrón de la molécula, queda una carga negativa remanente que en el caso del eteno se deslocaliza más fácilmente en el enlace π y σ que en el enlace σ simple que existe en un alcano. De todas formas, su acidez es menor que la de los alcoholes o los
ácidos carboxílicos
- Estabilidades relativas de los alquenos:
La hidrogenación de un alqueno da lugar al alcano correspondiente, por adición de hidrógeno al doble enlace. La energía desprendida depende de la estructura del alqueno y su medida nos da la idea de su estabilidad relativa:

| De los tres posibles butenos, el isómero trans es el que desprende menos calor de hidrogenación y, por tanto, debe ser el más estable. El 1-buteno es el menos estable porque es al que le corresponde la reacción más exotérmica. |
En general se observa que la estabilidad de un alqueno aumenta con la sustitución:
| Tetrasustituído > Trisustituído > Disustituído > Mononosustituído > Etileno |
Los isómeros trans son más estables que los correspondientes cis, porque aquéllos tienen menos interacciones estéricas desestabilizantes que éstos.
- Calores de formación:
La entalpía estándar de formación (H°f) o calor de formación de un compuesto es el cambio de calor que resulta de la formación de un mol de un compuesto a partir de sus elementos en sus estados estándar y se expresa en J/mol o en kJ/mol. El estado estándar, indicado con el superíndice (°), se refiere a la condición de 1 atm de presión. Si un elemento existe en más de una forma en condiciones estándar, se utiliza la forma más estable del elemento
para la reacción de formación.
- Reactividad de alquenos:
reacciones de adición:
| |
Un doble enlace C=C tiene una nube electrónica p desde la que se pueden ceder electrones a un atacante electrófilo. Por tanto, la reacción más importante de los alquenos es la adición electrófila.
La adición a alquenos es la reacción inversa a la eliminación:
Valores estimados para DHºde algunas reacciones de adición:
| X-Y |
DHºX-Y |
DHºC-X |
DHºC-Y |
DHº |
| H-H |
104 |
98 |
98 |
-27 |
| F-F |
37 |
110 |
110 |
-118 |
| Cl-Cl |
58 |
85 |
85 |
-47 |
| Br-Br |
46 |
71 |
71 |
-31 |
| I-I |
36 |
57 |
57 |
-13 |
| H-F |
128 |
98 |
110 |
-15 |
| H-Cl |
103 |
98 |
80 |
-10 |
| H-Br |
80 |
98 |
71 |
-24 |
| H-I |
64 |
98 |
57 |
-26 |
| H-OH |
119 |
98 |
92 |
-6 |
Las reacciones de adición son exotérmicas y, por tanto, son termodinámicamente favorables. Sin embargo, no se producen espontáneamente en general.
Por tanto, si existe un camino de reacción posible, es decir, con ET’s no demasiado altos en energía, las reacciones de adición se producirán con desprendimiento de energía.
|
- Adición de halógenos, agua y ácidos:
El cloro y el bromo se adicionan a alquenos para dar 1,2-dihaloalcanos. Por ejemplo, el 1,2-dicloroetano es sintetizado por la adición de cloro a eteno.

El F2 y el I2 no se emplean como reactivos en esta reacción. El Fluor reacciona de forma explosiva con alquenos y la adición de I2 es termodinamicamente desfavorable.
El mecanismo de esta reacción involucra la formación del ion bromonio en una primera etapa. En el segundo paso, el Br- actúa como nucleófilo abriendo el ciclo
del ion bromonio para formar un 1,2-dibromoalcano
- Funcionalización regioselectiva y estereoespecífica de alquenos.
-Hidrogenación catalítica de alquenos:
se refiere a la hidrogenación catalítica (usando Pt, Pd, o
Ni) formando alcanos del modo CH2=CH2 + H2 → CH3CH3.
- Productos anti- Markovnikov: Hidroboración y Adición de bromo radicalaria:
se refiere a la reacción con halógenos (representados por la X) del modo CH2=CH2 + X2 → XCH2CH2X. Por ejemplo, halogenación con
bromo.
-Oxidación de alquenos:
la oxidacion de un alqueno se puede mostrar facilmente ya que el doble enlace es nucleofilo, y el oxigeno tambien, asique necesitas un catalizador, como el h2o2, asi, se puede mostrar la oxidacion de un alqueno, mediante el proceso de ozonolisis, hoy en dia se ha comprobado que dentro del cuerpo humano se desarrolla este proceso.
- Reacciones de adición de los alquinos.
- Acidez de los alquinos terminales:
enlace carbono - hidrógeno de alquinos terminales está polarizado y muestra una ligera tendencia a ionizarse.
El Hidrógeno del propino es débilmente ácido, con un pKa = 25. Utilizando bases fuertes (NaH, LDA, NH2-) se puede desprotonar, obteniéndose su base
conjugada -propinil sodio- una especie muy básica y nucleófila.
- Sistemas conjugados: adición electrofílica y reacciones de cicloadición:
En mecanismos de reacción en química orgánica, una reacción de Adición Electrofílica (AEx o AdEx) es una reacción de adición donde en un compuesto químico, el sustrato de la reacción, se pierde un enlace pi para permitir la formación de dos nuevos enlaces sigma. En las reacciones de adición electrofílica, los sustratos más comunes tienen enlaces dobles o
enlaces triples carbono-carbono.
Y-Z + C=C → Y-C-C-Z.
Son aquellas reacciones en las que dos o más sistemas ð reaccionan para dar un ciclo con una insaturación menos.

6. Compuestos Aromáticos
-El benceno:
es un hidrocarburo aromático poliinsaturado de fórmula molecular C6H6, con forma de anillo (se le llama anillo bencénico, o aromático, ya que posee un olor característico) y puede considerarse una forma poliinsaturada del ciclohexano. En el benceno cada átomo de carbono ocupa el vértice de un hexágono regular, ocupa dos valencias con los dos átomos de
carbonos adyacentes, una tercera valencia con un átomo de hidrógeno y la cuarta denominada 'oculta' dirigiéndola hacia el centro del anillo hexagonal formada en algunos casos de carbono y en otros de alguna base nitrogenada. Cada átomo de carbono comparte su electrón libre con toda la molécula (según la teoría de orbitales moleculares), de modo que la estructura molecular adquiere una gran estabilidad y elasticidad. El benceno es un líquido incoloro y muy inflamable de aroma dulce, con un punto de fusión relativamente alto.

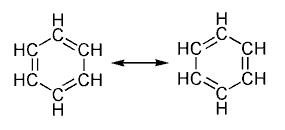
- Aromaticidad:
Es una propiedad de hidrocarburos cíclicosconjugados en la que los electrones de los enlaces dobles, libres de poder circular alrededor de un enlace a otro, sea enlace doble o simple, confieren a la
molécula una estabilidad mayor que la conferida si dichos electrones permanecieran fijos en el enlace doble.
- Modelo de resonancia:
Es un modelo de red neuronal artificial que basa su funcionamiento en la manera en que el cerebro procesa información y que describe una serie de modelos de
redes neuronales que utilizando métodos de aprendizaje supervisado y no supervisado abordan problemas tales como el reconocimiento y la predicción de patrones.

- Modelo orbital:
Un orbital atómico es una determinada solución particular, espacial e independiente del tiempo a la ecuación de Schrödinger para el caso de un electrón sometido a un
potencial coulombiano. La elección de tres números cuánticos en la solución general señalan unívocamente a un estado monoelectrónico posible.
Estos tres números cuánticos hacen referencia a la energía total del electrón, el
momento angular orbital y la proyección del mismo sobre el eje z del sistema del laboratorio y se denotan por

El nombre de orbital también atiende a la función de onda en representación de posición independiente del tiempo de un electrón en una
molécula. En este caso se utiliza el nombre orbital molecular.
La combinación de todos los orbitales atómicos dan lugar a la corteza electrónica representado por el modelo de capas electrónico. Este último se ajusta a los elemento según la
configuración electrónica correspondiente.

- Energía de resonancia en el benceno:
La Resonancia (denominado también Mesomería) en química es una herramienta empleada (predominantemente en química orgánica) para representar ciertos tipos de estructuras moleculares. La resonancia consiste en la combinación lineal de estructuras de una molécula (estructuras resonantes) que no coinciden con la estructura real, pero que mediante su combinación, nos acerca más a su estructura real. La resonancia molecular es un componente clave en la teoría del enlace covalente y su aparición crece cuando existen enlaces dobles o triples en la molécula, numerosos compuestos orgánicos presentan resonancia, como en el caso de los
compuestos aromáticos.
- Nomenclatura de bencenos sustituidos:
Los electrófilos también pueden reaccionar con anillos aromáticos sustituidos. La presencia de sustituyentes influye sobre dos aspectos de la sustitución electrófila aromática: la velocidad de reacción y la
regioselectividad de la misma.
- Otros anillos aromáticos y heteroaromáticos:
Los compuestos aromáticos pueden incluir heteroátomos en los anillos.

Piridina |

Pirimidina |

Pirazina |

Quinolina |

Isoquinolina |

Pirrol |

Indol |

Purina |

Imidazol |

Pirazol |

Furano |

Tiofeno |

Tiazol |
Los compuestos heteroaromáticos tienen nombres triviales. |
|
| Los sustituyentes se localizan numerando los anillos tal y como se indica. |
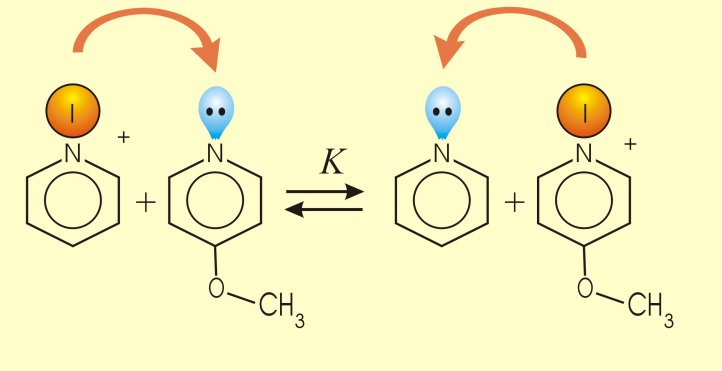
Los pares de electrones no compartidos de los heteroátomos pueden o no participar en los 4n+2 electrones, que son condición de aromaticidad, dependiendo de la estructura. En este sentido, ¿qué puedes deducir de los siguientes datos de basicidad?.
Protonación de la piridina

pKb = 8.8 |
La piridina es básica. Esto indica que el par de electrones de la piridina no está implicado en la aromaticidad del anillo. Los electrones p por sí sólos cumplen la regla de Hückel de los 4n+2 (n=1) electrones. |

pKb = 18.4 |
|
|
| El pirrol es muy poco básico. Se comprueba que, de protonarse en un medio de fortaleza ácida suficientemente alto, la protonación ni siquiera ocurre en el nitrógeno. Los electrones del nitrógeno del pirrol están totalmente comprometidos en el sextete aromático y resulta muy costoso energéticamente compartirlos hacia un atacante ácido. |
|
La estructura electrónica de ambos anillos nos permite comprender sus propiedades de basicidad diferentes:

La piridina es básica |
|

El pirrol no es básico |
|
| ¿Qué nitrógenos son básicos y cuéles no en los heterociclos indicados al principio de esta página? Respuesta |
En cuanto a la reactividad, ¿son la piridina y el pirrol más o menos reactivos que el benceno?
 |
El nitrógeno de la piridina retira carga del anillo aromático. La piridina es menos reactiva que el benceno frente a la SEAr. |
 |
|
|
El nitrógeno del pirrol no puede retirar carga del anillo. En todo caso puede cederla.
El pirrol es más reactivo que el benceno frente a la SEAr. |
|
- sustitucion electrofilica aromatica:
La halogenación aromática con bromo o cloro conduce a los correspondientes haluros de arilo usando como
catalizadores haluros de hierro (FeX3) o de aluminio (AlX3).
La sulfonación aromática del benceno con
ácido sulfúrico fumante produce ácido bencenosulfónico.
La reacción de Friedel-Crafts en sus dos versiones de alquilación y acilación. Teniendo como reactivos respectivamente haluros de alquilo o haluros de acilo (también conocidos como haluros de ácido o haluros de alcanoílo). Habitualmente el cloruro de aluminio (AlCl3) es el
catalizador.
Ejemplo de acilación de Friedel-Crafts. Aunque no aparece, requiere tratamiento acuoso final.
Ejemplo de alquilación de Friedel-Crafts.
Otras sustituciones
Otras reacciones que siguen un esquema de sustitución electrófila aromática son un grupo de reacciones de formilación aromática que incluyen la reacción de Vilsmeier-Haack, la reacción de Gattermann Koch y la
reacción de Reimer-Tiemann.
Otras reacciones son el acomplamiento diazoico donde el electrófilo son sales de diazonio aromáticas, la reacción de Kolbe-Schmitt en la que el electrófilo es dióxido de carbono y la condensación de Pechmann donde el electrófilo es un
grupo carbonilo activado.
En la
reacción de Lehmstedt-Tanasescu, compuesta de varias etapas, uno de los electrófilos es un intermedio N-nitroso (N-NO).
- Halogenación del benceno:
El benceno reacciona con halógenos en presencia de ácidos de Lewis para formar derivados halogenados.
La cloración se puede llevar a cabo de forma similar a la bromación. La reacción con flúor y yodo se realiza muy poco frecuentemente. En el caso del flúor la reacción es dificil de controlar por su elevada reactividad. Por el contrario, el yodo reacciona lentamente y tiene un equilibrio desfavorable.
-Necesidad de catalizador:
Un catalizador es una sustancia química, simple o compuesta, que modifica la velocidad de una reacción química, interviniendo en ella pero sin llegar a formar parte de los productos resultantes de la misma. Los catalizadores se caracterizan con arreglo a las dos variables principales que los definen: la fase activa y la selectividad. La actividad y la selectividad, e incluso la vida misma del catalizador, depende directamente de la fase activa utilizada, por lo que se distinguen dos grandes subgrupos: los elementos y compuestos con propiedades de conductores electrónicos y los compuestos que carecen de electrones libres y son, por lo tanto, aislantes o dieléctricos. La mayoría de los catalizadores sólidos son los metales o los óxidos, sulfuros y haloideos de elementos metálicos y de semimetálicos como los elementos boro aluminio, y silicio. Los catalizadores gaseosos y líquidos se usan usualmente en su forma pura o en la combinación con solventes o transportadores apropiados; los catalizadores sólidos se dispersan usualmente en otras sustancias conocidas como apoyos de catalizador Un catalizador en disolución con los reactivos, o en la misma fase que ellos, se llaman catalizador homogéneo. El catalizador se combina con uno de los reactivos formando un compuesto intermedio que reacciona con el otro más fácilmente. Sin embargo, el catalizador no influye en el equilibrio de la reacción, porque la descomposición de los productos en los reactivos es acelerada en un grado similar.
Nitración y sulfonación del benceno:
La nitración del benceno puede llevarse a cabo mezclando ácido nítrico con benceno, en presencia de ácido sulfúrico concentrado, y calentando la mezcla:
Esta reacción es una sustitución electrófilica análoga a las halogenaciones recién discutidas. El agente electrófilico es el NO2; los ácidos nítrico y sulfúrico se limitan a producir el NO2+.
La sustitución electrófila comienza cuando el ácido sulfúrico, por ser extraordinariamente fuerte, protona al nítrico dando H2N03+ que puede perder agua para formar NO2+. El ácido sulfúrico facilita esta ultima reacción capturando el agua formada. Entonces el ion NO2+ ataca al anillo del benceno y después ocurren los pasos ya conocidos. Podemos formular el mecanismo completo así:
SULFUNACIÓN
El benceno reacciona lentamente con el ácido sulfúrico a temperaturas elevadas. El producto de la reacción es el ácido bencenosulfónico:
- Formación de enlaces C-C:
Un enlace carbono-carbono es un enlacecovalente entre dos átomos de carbono. La forma más común es el enlace simple - un enlace compuesto por dos electrones, uno de cada uno de los dos átomos. El enlace simple carbono-carbono es un enlace sigma y se dice que se forma entre un orbital híbrido de cada uno de los átomos de carbono. En el etano, los orbitales son sp3, pero
también pueden existir enlaces simples formados por átomos de carbono con otras hibridaciones (por ejemplo, sp2 a sp2). En efecto, los átomos de carbono en
el enlace simple no necesitan ser de la misma hibridación. Los átomos de carbono también pueden formar enlace doble, constituyendo alquenos, o enlace triple, en alquinos. Un enlace doble está formado con un orbital híbrido sp2 y un
orbital p que no está involucrado en la hibridación. Un enlace triple está formado con un orbital híbrido sp y dos orbitales p de cada átomo. El uso de los orbitales p forma un
enlace pi.
El carbono tiene la característica única entre todos los elementos de formar cadenas largas y estables de sus propios átomos, una propiedad llamada catenación. Esto, junto con la fuerza del enlace carbono-carbono da origen a un número enorme de formas moleculares, muchas de las cuales son importantes elementos estructurales de la vida, así los compuestos de carbono tienen su propio campo de estudio: la
química orgánica.
-Las reacciones de Friedel-Crafts:
Las reacciones de Friedel-Crafts son un tipo de reacción de sustitución electrófila aromática en las que en un compuesto aromático un átomo de hidrógeno es sustituido por un alquilo, alquilación de Friedel-Crafts, o un grupo acilo, acilación de Friedel-Crafts. Fueron descubiertas el año 1877 por el químico francés Charles Friedel y por el químico americano
James M. Crafts.
- Activación y desactivación del anillo bencénico:
Los sustituyentes presentes sobre un compuesto aromático influyen fuertemente en su reactividad. Estos sustituyentes o grupos se clasifican en: activantes o desactivantes. Los primeros provocan que la reacción sea más rápida que en el compuesto no sustituido y los segundos lo contrario. Un grupo o sustituyente será activante si cede densidad electrónica al sistema aromático o desactivante si en cambio atrae electrones.
Grupos activantes
Un
grupo activante es aquel cuya presencia aumenta la reactividad, la velocidad de reacción, del anillo aromático frente a la sustitución electrófila aromática respecto a cuando ese grupo está ausente. La introducción de un grupo activante en un compuesto aromático no sustituido conducirá frecuentemente a una polisustitución.
Este aumento de la velocidad de reacción se debe a que estos grupos estabilizan el intermedio catiónico formado durante la sustitución a través de la cesión de densidad electrónica sobre el sistema anular, ya sea por efecto inductivo o por efecto resonante (o mesomero). Esto implica que la barrera o
energía de activación disminuya para la primera étapa de la reacción, que es la que controla la velocidad global de la misma.
- Activación por efecto inductivo:
Los alquilos son activantes débiles por efecto inductivo (y también por hiperconjugación). El efecto inductivo está controlado por la electronegatividad. Un ejemplo de un anillo aromático débilmente activado por un sustityente alquilo es el
tolueno.
- Activación donde predomina el efecto resonante:
Los grupos que pueden ceder por resonancia pares de electrones no compartidos al sistema π son activantes. Grupos activantes por resonancia son las funciones amino,
hidroxi y sus derivados.
Anilina, ejemplo de anillo aromático fuertemente activado
En la anilina el grupo amino, dada la mayor electronegatividad del átomo de nitrógeno respecto al de carbono, es atrayente de electrones por efecto inductivo. En cambio por efecto resonante es dador de electrones, ya que tiene un par de electrones sin compartir que pueden deslocalizarse por el anillo aromático. En este caso el efecto resonante domina sobre el inductivo, y el efecto global es que la anilina está fuertemente activada frente a la sustitución electrófila aromática. Otro ejemplo sería el fenol.
Grupos desactivantes
un grupo desactivante es aquel cuya presencia disminuye la reactividad, la velocidad de reacción, del anillo aromático frente a la sustitución electrófila aromática respecto a cuando ese grupo está ausente. Por tanto la introducción de un grupo desactivante en un compuesto aromático no sustituido hará más difícil, condiciones más agresivas, una segunda sustitución.
Esta disminución de la velocidad de reacción se debe a que estos grupos desestabilizan el intermedio catiónico. Esto es así debido a que son grupos que retiran densidad electrónica del sistema aromático, ya sea por efecto inductivo o por efecto resonante. Esto supone que la barrera o
energía de activación de la primera etapa se eleve, y por tanto disminuya la reactividad.
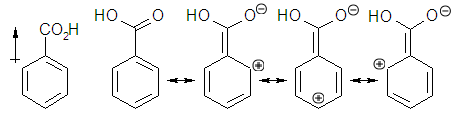
Acido benzoico, ejemplo de anillo muy desactivado.
Otros ejemplo de compuestos fuertemente desactivados son el nitrobenceno, el benzaldehído o el (trifluorometil)-benceno (
Ph-CF3).
Los halobencenos, (Ph-X donde X = F, Cl, Br o I), dada la electronegatividad de los
halógenos, están ligeramente desactivados por efecto inductivo.
-Orientación en la sustitución aromática electrofílica:
La presencia de dos sustituyentes en un anillo complica el problema de la orientación, pero aun así frecuentemente pueden hacerse pronósticos muy definidos. En primer lugar, ambos sustituyentes pueden estar ubicados de modo tal, que la influencia directora de uno refuerce la del otro; por ejemplo, en I, II y III, la orientación debe ser claramente la indicada por las flechas. Por otra parte, cuando el efecto director de un grupo es opuesto al del otro, puede ser difícil predecir el producto principal. En tales casos, a menudo se obtienen mezclas complejas de varios compuestos.
- Teoría de la orientación en la sustitución electrófila aromática:
Antes de justificar la orientación en la sustitución electrofílica, examinemos los hechos.
Un grupo activante activa todas las posiciones del anillo bencénico: incluso las meta son más reactivas que cualquier otra posición individual del propio benceno. Dirige a orto y para porque las activas más que a la meta.
Un grupo desactivante desactiva todas las posiciones del anillo, incluso las meta. Dirige meta simplemente, porque desactiva las posiciones orto y para más que a las meta.
Así, tanto la orientación orto-para como la meta surgen de la misma forma: el efecto de cualquier grupo sea activante o desactivante es más intenso en las posiciones orto y para.
Para apreciar si esto es lo esperado, comparemos, por ejemplo, los carbocationes generados por el ataque en las posiciones para y meta del tolueno, un compuesto con un grupo activante. Cada uno de ellos es un híbrido de tres estructuras: I-III para para, IV-VI para meta. En una de estas seis estructuras, la II, la carga positiva se encuentra ubicada en el carbono que tiene el –CH3. Aunque este libera electrones para todas las posiciones del anillo, lo hace más intensamente hacia el carbono más cercano. Por consiguiente, la estructura II es una particularmente estable. Debido a la contribución de esta estructura II, el carbocatión híbrido que resulta del ataque a la posición para es más estable que el resultante del ataque a la posición meta. Por tanto, la sustitución es más rápida en para que en meta.
- Efectos de varios sustituyentes:
Como el benceno, el tolueno experimenta sustitución electrofílica aromática: sulfonación, por ejemplo, Aunque son posibles tres productos monosulfonados, en realidad esta reacción sólo da cantidades apreciables de dos de ellos: los isómeros orto y para. El benceno y el tolueno son insolubles en ácido sulfúrico, mientras que los ácidos sulfónicos son muy solubles. La desaparición de la capa de hidrocarburo indica que se ha completado la reacción. Al ser agitado a temperatura ambiente con ácido sulfúrico fumante, el benceno reacciona completamente entre 20 y 30 minutos, mientras que el tolueno lo hace entre uno y dos minutos.
El estudio de la nitración, halogenación y alquilación de Friedel-Crafts da resultados análogos. De alguna manera, el grupo alquilo hace más reactivo al anillo bencénico y que el benceno no sustituido, y dirige el reactivo ataque a las posiciones anulares orto y para.
Por otra parte, y para considerar un ejemplo diferente, se ha encontrado que el nitrobenceno se sutituye de forma más lenta que el benceno, y que produce principalmente el isómero meta.
Como el metilo o el nitro, cualquier grupo unido a un anillo bencénico lo afecta en su reactividad y determina la orientación de la sustitución. Cuando un reactivo electrófilo ataca un anillo aromático, el grupo ya enlazado determina lo fácil que será el ataque, y dónde sucederá.
Cuando un grupo hace que un anillo sea más reactivo que el benceno, se llama grupo activante; si produce el resultado contrario, se conoce como grupo desactivante.
Un grupo que motiva un ataque en las posiciones orto y para es un director orto-para; uno que ocasiona lo mismo en las posiciones meta, se denomina director meta.
En este capítulo examinaremos los métodos que se emplean para medir estos efectos sobre la reactividad y la orientación, sus resultados y una teoría que justifica dichos resultados. Evidentemente, la teoría se basa en el mecanismo más probable para la sustitución electrofílica aromática. Veremos cuál es este mecanismo y, también, algunas pruebas que lo apoyan. Primero examinemos los hechos.
- Hidrocarburos aromáticos policíclicos:
Un hidrocarburo aromático policíclico (HAP o PAH, por sus siglas en inglés) es un compuesto químico que se compone de anillos aromáticos simples que se han unido, y no contiene heteroátomos ni lleva sustituyentes.[1] Los HAPs se
encuentran en el petróleo, el carbón y en depósitos de alquitrán y también como productos de la utilización de combustibles (ya sean fósiles o biomasa). Como contaminantes han despertado preocupación debido a que algunos compuestos han sido identificados como carcinógenos, mutágenos y
teratógenos.
También se encuentran en el medio interestelar, en cometas y en meteoritos, y son candidatos a moléculas básicas en el origen de la vida. En el
grafeno el motivo HAP se extiende en grandes láminas bidimensionales.
- Reactividad del sistema bencílico:
Sistema dirigido por un compuesto organico.

7. Compuestos orgánicos Halgenados
-Estructura de los haluros de alquilo.
- Propiedades físicas:
Debido a su mayor peso molecular, los haloalcanos tiene puntos de ebullición considerablemente más altos que los alcanos de igual número de carbonos (Tabla 1). Para un grupo alquilo dado, el punto de ebullición aumenta con el incremento en el peso atómico del halógeno, de modo que un fluoruro hierve a la temperatura más baja, y un yoduro, a la más elevada.
Para un halógeno determinado, el punto de ebullición aumenta al aumentar el número de carbonos: al igual que en los alcanos, el aumento de punto de ebullición es de unos 20-30 grados por cada carbono adicional, salvo en el caso de los homólogos muy pequeños. Igual que antes, el punto de ebullición disminuye con el aumento de las ramificaciones ya impliquen un grupos alquilo o al propio halógeno.
-Sustitución nucleofílica:
Los componentes requeridos para la sustitución nucleofílica son: sustrato, nucleófilo y disolvente. El sustrato consta de dos partes: grupo alquilo y grupo saliente. Nos ocuparemos del grupo alquilo durante gran parte de este capítulo; estudiaremos las funciones del disolvente en el capítulo 6. Por el momento examinaremos los otros componentes de estos sistemas: los nucleófilos y grupos salientes.
- Aplicaciones sintéticas.
-Mecanismo SN2:
La SN2 (sustitución nucleófila bimolecular) es una reacción concertada, es decir, transcurre en una única etapa.
El mecanismo consiste en el ataque del nucleófilo al carbono que contiene el grupo saliente. Este carbono presenta una polaridad positiva importante, debida a la electronegatividad del halógeno. Al mismo tiempo que ataca el nucleófilo se produce la ruptura del enlace carbono-halógeno, obteniéndose el producto final.
- Mecanismo SN1: Solvolisis:
La SN1 tiene un mecanismo por etapas. En el primer paso se ioniza el sustrato por pérdida del grupo saliente, sin que el nucleófilo actúe, formándose un carbocatión. En el segundo paso el nucleófilo ataca al carbocatión formado, obteniéndose el producto final.
Etapa 1. Disociación del sustrato, para formar el carbocatión. Es el paso lento de la reacción.
Etapa 2. Ataque del nucleófilo al carbocatión formado.
Etapa 3. Desprotonación del agua para formar el alcohol
La etapa lenta del mecanismo es la formación del carbocatión, dependiendo la velocidad exclusivamente del sustrato.
-Estabilidad de los carbocationes:
La estabilidad de los carbocationes se incrementa con el número de grupos alquilo unidos al átomo de carbono que lleva la carga. Los carbocationes terciarios son más estables (y se forman más rápidamente) que los carbocationes secundarios; los carbocationes primarios son altamente inestable porque, mientras los carbocationes de orden mayor están estabilizados por hiperconjugación y por impedimento estérico, los carbocationes primarios no lo están. En consecuencia, reacciones como la reacción SN1 y la reacción de eliminación E1 normalmente no ocurren si involucrasen a un carbocatión primario. Una excepción a esto ocurre cuando hay un enlace doble carbono-carbono en la posición 2 respecto al átomo de carbono ionizado. Tales cationes, como el catión alilo CH2=CH-CH2+ y el catión
bencilo C6H5-CH2+ son más
estables que los otros carbocationes. Las moléculas que pueden formar carbocationes alilo o bencilo son altamente reactivas.
Los carbocationes sufrenreacciones de reordenamientode estructuras menos estables a estructuras igualmente estables o más estables con
constante de velocidadmayor a 1.0E9 s-1. Este hecho complica las trayectorias de síntesis de
muchos compuestos. Por ejemplo, cuando el 3-pentanol se caliente con HCl
acuoso, el carbocatión 3-pentilo formado se transpone a una mezcla estadística de 3-pentil y 2-pentil. Estos cationes reaccionan con ion cloruro para producir cerca de 1/3 de 3-cloropentano y 2/3 de 2-cloropetano.
-Comparación entre mecanismo unimolecular y bimolecular:
La eliminación bimolecular o E2 consiste en un mecanismo concertado de abstracción de un protón por parte de una base fuerte y la salida simultánea de un grupo saliente situado en β, en el
carbono contiguo, formándose una insaturación (doble enlace).

La eliminación unimolecular o E1 tiene lugar sobre derivados alquílicos secundarios o terciarios según un mecanismo de dos étapas. En la primera se produce la salida del grupo saliente para formar el
carbocatión y a continuación la pérdida de un protón en β para formar un doble enlace.
-Deshidrohalogenación: Reacción de eliminación:
Enquímica orgánica, una reacción de eliminación es el proceso inverso a unareacción de adición. Es unareacciónorgánica en la que dossustituyentesson eliminados de unamolécula, creándose también unainsaturación, ya sea un doble o triple enlace, o un anillo. En el caso particular de que los dos grupos sean eliminados de un mismo centro el resultado sería un
carbeno:CR2.
Las reacciones de eliminación más importantes son aquellas en las que los dos grupos que se eliminan están situados en átomos adyacentes, dando lugar a una nueva insaturación en la forma de un alqueno, un alquino o un
carbonilo.
- Mecanismos E1 y E2:
La eliminación unimolecular o E1 tiene lugar sobre derivados alquílicos secundarios o terciarios según un mecanismo de dos étapas. En la primera se produce la salida del grupo saliente para formar el
carbocatióny a continuación la pérdida de un protón en β para formar un doble enlace.